

 Cart
Cart
 sales@molnova.com
sales@molnova.com


Relugolix
CAS No. 737789-87-6
Relugolix( TAK385 | TAK-385 | TAK 385 | Relugolix )
Catalog No. M17586 CAS No. 737789-87-6
Relugolix is an orally available, non-peptide gonadotropin-releasing hormone (GnRH or luteinizing hormone-releasing hormone (LHRH)) antagonist, with potential antineoplastic activity.
Purity : >98% (HPLC)
 COA
Datasheet
HNMR
HPLC
MSDS
Handing Instructions
COA
Datasheet
HNMR
HPLC
MSDS
Handing Instructions
| Size | Price / USD | Stock | Quantity |
| 5MG | 65 | In Stock |


|
| 10MG | 106 | In Stock |
|
| 25MG | 188 | In Stock |
|
| 50MG | 312 | In Stock |
|
| 100MG | 484 | In Stock |
|
| 500MG | 1035 | In Stock |
|
| 1G | Get Quote | In Stock |
|
Biological Information
-
Product NameRelugolix
-
NoteResearch use only, not for human use.
-
Brief DescriptionRelugolix is an orally available, non-peptide gonadotropin-releasing hormone (GnRH or luteinizing hormone-releasing hormone (LHRH)) antagonist, with potential antineoplastic activity.
-
DescriptionRelugolix, also known as TAK-385, is a luteinizing hormone-releasing hormone (LH-RH) receptor antagonist administered orally. By preventing LH-RH from binding with the LH-RH receptor in the anterior pituitary gland and suppressing the secretion of luteinizing hormone (LH) and follicle stimulation hormone (FSH) from the anterior pituitary gland, TAK-385 controls the effect of LH and FSH on the ovary, reduces the level of estrogen in blood, which is known to be associated with the development of endometriosis and uterine fibroids, and is expected to improve the symptoms of these disorders.(In Vitro):Relugolix exhibits strong binding affinity (IC50=0.32 nM) for the monkey receptor comparable to that for the human receptor (IC50=0.33 nM) while displaying a 30000-fold decrease for the rat receptor (IC50=9800 nM). The antagonistic in vitro activity of TAK-385 with respect to the human receptor (IC90=18 nM) exceeded that for the monkey receptor (IC90=1700 nM) by 95-fold in the presence of 40% serum.(In Vivo):Relugolix (oral administration; 1-3 mg/kg; single dose for pharmacokinetic study) exhibits a good pharmacokinetic profile and obvious suppressive effects of circulating LH levels in monkeys at a dose of 1 mg/kg. The pharmacokinetic profile exhibits with 16.0 ng/mL, 2.7 h, and 90.1 ng for Cmax, Tmax, and AUCo, respectively in male cynomolgus monkeys.Relugolix (oral administration; 3, 10 or 30 mg/kg; twice daily; 4 weeks) significantly decreases the testis weight, and reduces the ventral prostate weight at 3 mg/kg and decreases it to castrate levels at 10 mg/kg in male hGNRHR-knock-in mice.Relugolix (oral administration; 30, 100 or 200 mg/kg; twice daily; 4 weeks) induces constant diestrous phases in all mice within the first week at 100 mg/kg, and significantly decreases the weights of ovaries and uteri at this dose after 4 weeks in female hGNRHR-knock-in mice.
-
In VitroRelugolix exhibits strong binding affinity (IC50=0.32 nM) for the monkey receptor comparable to that for the human receptor (IC50=0.33 nM) while displaying a 30000-fold decrease for the rat receptor (IC50=9800 nM). The antagonistic in vitro activity of TAK-385 with respect to the human receptor (IC90=18 nM) exceeded that for the monkey receptor (IC90=1700 nM) by 95-fold in the presence of 40% serum.
-
In VivoRelugolix (oral administration; 1-3 mg/kg; single dose for pharmacokinetic study) exhibits a good pharmacokinetic profile and obvious suppressive effects of circulating LH levels in monkeys at a dose of 1 mg/kg. The pharmacokinetic profile exhibits with 16.0 ng/mL, 2.7 h, and 90.1 ng for Cmax, Tmax, and AUCo, respectively in male cynomolgus monkeys.Relugolix (oral administration; 3, 10 or 30 mg/kg; twice daily; 4 weeks) significantly decreases the testis weight, and reduces the ventral prostate weight at 3 mg/kg and decreases it to castrate levels at 10 mg/kg in male hGNRHR-knock-in mice.Relugolix (oral administration; 30, 100 or 200 mg/kg; twice daily; 4 weeks) induces constant diestrous phases in all mice within the first week at 100 mg/kg, and significantly decreases the weights of ovaries and uteri at this dose after 4 weeks in female hGNRHR-knock-in mice. Animal Model:Male hGNRHR-knock-in mice Dosage:3, 10 or 30 mg/kg Administration:Oral administration; 3, 10 or 30 mg/kg; twice daily; 4 weeksResult:Decreased testicular function.Animal Model:Female hGNRHR-knock-in miceDosage:30, 100 or 200 mg/kg Administration:Oral administration; 30, 100 or 200 mg/kg; twice daily; 4 weeks Result:Suppressed the hypothalamic–pituitary–gonadal axis to gonadectomized levels.Downregulated GnRH receptor mRNA levels in the pituitary.
-
SynonymsTAK385 | TAK-385 | TAK 385 | Relugolix
-
PathwayApoptosis
-
TargetMDM2-p53
-
RecptorGNRHR
-
Research AreaCancer
-
Indication——
Chemical Information
-
CAS Number737789-87-6
-
Formula Weight623.63
-
Molecular FormulaC29H27F2N7O5S
-
Purity>98% (HPLC)
-
SolubilityDMSO : 50 mg/mL. 80.18 mM;
-
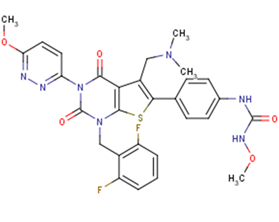
SMILESCN(C)Cc1c(sc2c1c(=O)n(c(=O)n2Cc1c(cccc1F)F)c1nnc(cc1)OC)c1ccc(cc1)NC(=O)NOC
-
Chemical Name1-(4-(1-(2,6-difluorobenzyl)-5-((dimethylamino)methyl)-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl)phenyl)-3-methoxyurea
Shipping & Storage Information
-
Storage(-20℃)
-
ShippingWith Ice Pack
-
Stability≥ 2 years
Reference
1. MacLean DB et al. J Clin Endocrinol Metab. 2015 Dec;100(12):4579-87.
molnova catalog



related products
-
BI-0252
A potent, highly selective, orally active MDM2-p53 interaction inhibitor with IC50 of 4 nM.
-
Pifithrin-β hydrobro...
A cyclic analog of Pifithrin-α and a small molecule inhibitor of p53; prevents dexamethasone-induced cell death in murine thymocytes with EC50 of 2.01 uM.
-
COTI-2
COTI-2 (COTI2) is an orally available thiosemicarbazone that may act on mutant forms of p53 and PI3K/AKT/mTOR pathway.
