

 Cart
Cart
 sales@molnova.com
sales@molnova.com


PRN1371
CAS No. 1802929-43-6
PRN1371( —— )
Catalog No. M22475 CAS No. 1802929-43-6
PRN1371 is a specific and potent FGFR1-4 and CSF1R inhibitor (IC50s: 0.6/1.3/4.1/19.3/8.1 nM for FGFR1/2/3/4 and CSF1R).
Purity : >98% (HPLC)
 COA
Datasheet
HNMR
HPLC
MSDS
Handing Instructions
COA
Datasheet
HNMR
HPLC
MSDS
Handing Instructions
| Size | Price / USD | Stock | Quantity |
| 5MG | 160 | In Stock |


|
| 10MG | 290 | In Stock |
|
| 25MG | 492 | In Stock |
|
| 50MG | 709 | In Stock |
|
| 100MG | 972 | In Stock |
|
| 200MG | Get Quote | In Stock |
|
| 500MG | Get Quote | In Stock |
|
| 1G | Get Quote | In Stock |
|
Biological Information
-
Product NamePRN1371
-
NoteResearch use only, not for human use.
-
Brief DescriptionPRN1371 is a specific and potent FGFR1-4 and CSF1R inhibitor (IC50s: 0.6/1.3/4.1/19.3/8.1 nM for FGFR1/2/3/4 and CSF1R).
-
DescriptionPRN1371 is a specific and potent FGFR1-4 and CSF1R inhibitor (IC50s: 0.6/1.3/4.1/19.3/8.1 nM for FGFR1/2/3/4 and CSF1R).PRN1371 presents a unique profile of high biochemical and cellular potency (FGFR1 IC50:0.6 nM, SNU16 IC50: 2.6 nM), prolonged target engagement (FGFR1 occupancy 24 h=96%), < 30% hERG inhibition at 1 μM, and good predicted ADME stability with BME reactivity Kd>100 μM.In PK studies of rat, dog, and cynomolgus monkey, PRN1371show rapid iv clearance in all species. PRN1371 shows rapid clearance (Cl=160 mL per min per kg), yet dosing p.o (20 mg/kg) demonstrates high oral exposure (AUC=4348 h·ng/mL) and a reasonable half-life (t1/2=3.8 h). Low levels of pFGFR2 confirm the ability of PRN1371 to block FGFR2 activity in tumor tissue. PRN1371 induces a dose-dependent reduction in tumor volume and up to 68% tumor growth inhibition at the highest dose of 10 mg/kg b.i.d. following 27 days of treatment. All doses are well tolerated with no significant bodyweight loss.(In Vitro):PRN1371 presents a unique profile of high biochemical and cellular potency (FGFR1 IC50=0.6 nM, SNU16 IC50=2.6 nM), prolonged target engagement (FGFR1 occupancy 24 h=96%), < 30% hERG inhibition at 1 μM, and good predicted ADME stability with BME reactivity Kd>100 μM. Broader kinome-wide biochemical profiling of PRN1371 against 251 kinases identifies only FGFR1?4 and CSF1R as being potently inhibited.(In Vivo):PK studies of PRN1371 in rat, dog, and cynomolgus monkey show rapid iv clearance in all species. PRN1371 shows rapid clearance (Cl=160 mL per min per kg), yet dosing po (20 mg/kg) demonstrates high oral exposure (AUC=4348 h·ng/mL) and a reasonable half-life (t1/2=3.8 h). Low levels of pFGFR2 confirms the ability of PRN1371 to block FGFR2 activity in tumor tissue. PRN1371 induces a dose-dependent reduction in tumor volume and up to 68% tumor growth inhibition at the highest dose of 10 mg/kg b.i.d. following 27 days of treatment. All doses are well tolerated with no significant body weight loss. PRN1371 free base has been administered orally once daily as powder in a capsule on a 28-day continuous schedule. Human plasma concentrations for doses ranging from 15 to 35 mg confirm good oral exposure, rapid systemic clearance, no accumulation from day 1 to day 15, and a dose-dependent increase in AUC. Serum phosphate, a pharmacodynamic marker of FGFR inhibition, is increased for all doses studied and shows a dose-dependent increase between 20 and 35 mg, despite the administration of prophylactic phosphate binders.
-
In VitroPRN1371 presents a unique profile of high biochemical and cellular potency (FGFR1 IC50=0.6 nM, SNU16 IC50=2.6 nM), prolonged target engagement (FGFR1 occupancy 24 h=96%), < 30% hERG inhibition at 1 μM, and good predicted ADME stability with BME reactivity Kd>100 μM. Broader kinome-wide biochemical profiling of PRN1371 against 251 kinases identifies only FGFR1?4 and CSF1R as being potently inhibited.
-
In VivoPK studies of PRN1371 in rat, dog, and cynomolgus monkey show rapid iv clearance in all species. PRN1371 shows rapid clearance (Cl=160 mL per min per kg), yet dosing po (20 mg/kg) demonstrates high oral exposure (AUC=4348 h·ng/mL) and a reasonable half-life (t1/2=3.8 h). Low levels of pFGFR2 confirms the ability of PRN1371 to block FGFR2 activity in tumor tissue. PRN1371 induces a dose-dependent reduction in tumor volume and up to 68% tumor growth inhibition at the highest dose of 10 mg/kg b.i.d. following 27 days of treatment. All doses are well tolerated with no significant body weight loss. PRN1371 free base has been administered orally once daily as powder in a capsule on a 28-day continuous schedule. Human plasma concentrations for doses ranging from 15 to 35 mg confirm good oral exposure, rapid systemic clearance, no accumulation from day 1 to day 15, and a dose-dependent increase in AUC. Serum phosphate, a pharmacodynamic marker of FGFR inhibition, is increased for all doses studied and shows a dose-dependent increase between 20 and 35 mg, despite the administration of prophylactic phosphate binders.
-
Synonyms——
-
PathwayAngiogenesis
-
TargetFGFR
-
RecptorFGFR1| FGFR2| FGFR3| FGFR4| CSF-1R
-
Research AreaCancer
-
IndicationSolid tumours; Urogenital cancer
Chemical Information
-
CAS Number1802929-43-6
-
Formula Weight561.46
-
Molecular FormulaC26H30Cl2N6O4
-
Purity>98% (HPLC)
-
SolubilityDMSO:12 mg/mL (21.38 mM; Need ultrasonic and warming)
-
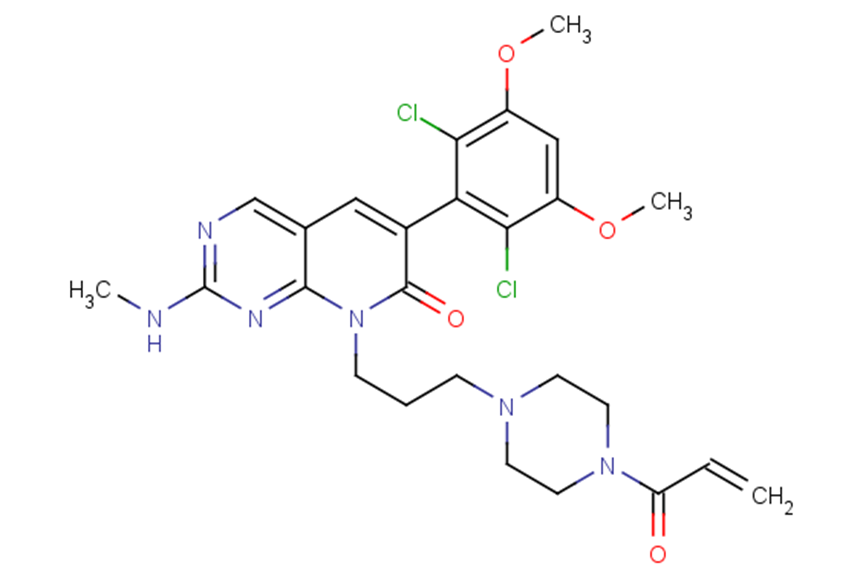
SMILESCNc1ncc2cc(-c3c(Cl)c(OC)cc(OC)c3Cl)c(=O)n(CCCN3CCN(CC3)C(=O)C=C)c2n1
-
Chemical Name——
Shipping & Storage Information
-
Storage(-20℃)
-
ShippingWith Ice Pack
-
Stability≥ 2 years
Reference
1.Brameld KA, et al. Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (PRN1371) for the Treatment of Solid Tumors. J Med Chem. 2017 Aug 10;60(15):6516-6527.
molnova catalog



related products
-
Vofatamab
Vofatamab (B-701) is a fully human monoclonal antibody targeting FGFR3. Vofatamab has potential anticancer and antitumor activity and is often used in combination with other compounds to treat cancer.
-
SM27
SM27 is a fibroblast growth factor 2 (FGF2) inhibitor with anti-angiogenic activity and can be used to study tumours.
-
H3B-6527
A potent, highly selective covalent FGFR4 inhibitor with IC50 of <1.2 nM.
